黄 响1 ,陈林东1 ,余国枢2 (1. 茂名环星新材料股份有限公司,茂名;2. 江门市质量计量监督检测所,江门)
摘 要:根据炭黑中多环芳烃的检测方法,参照国标GB/ T29614-2013和GB/T3780.28检测了多环芳烃含量。总结了影
响多环芳烃检测结果的几种因素,如仪器状态、进样针、提纯净化方式以及色谱柱的选用等,并针对这些因素总结经验制
定对策,以提高气质联用法检测其含量的分析准确度。
关键词:气质联用仪;多环芳烃;分析准确度
引 言
多环芳烃(Polycyclic Aromatic Hydrocarbons, PAHs)是一类含有两个以上苯环以稠环式相连所
组成的碳氢化合物,是数量最多的、重要的一类环 境致癌物质[1]。PAHs 具有水性、半挥发性、难降 解性的特点,具有强烈致癌、致畸、致突变性和生
殖毒性,因此倍受到国际上的广泛的关注[2-4]。欧 盟2005/69/EC指令和REACH法规对轮胎中的多环 芳烃含量做出了限制要求,我国出口的轮胎面临
着技术壁垒和挑战。
轮胎中的多环芳烃主要来自炭黑与操作油[5]。 因此,分析检测和控制炭黑中的多环芳烃含量,生 产出低多环芳烃的环保炭黑具有重要的意义。目
前检测炭黑中的PAHs含量尚未有实行的国家标准 和规范,国内对炭黑中PAHs含量检测主要的参考 标准有《GB/T29614-2013 硫化橡胶中多环芳烃含
量的测定》,该标准方法B气相色谱-质谱法提到 的检测原理是:试样经超声波水浴提取,提取液冷 却后即为待测溶液,用气相色谱 - 质谱联用仪
(GC-MS)测定,内标法定量。本文参照该标准以 及《GB/T3780.28炭黑 第28部分:多环芳烃含量的 测定(征求意见稿)》,结合平常经验总结和探讨
了影响炭黑中多环芳烃含量分析测准确度的几个 因素,以期提高分析准确度。
1 实验部分
1.1 主要仪器
GCMS-QP2010 SE GC/MS联用仪,日本岛津公 司;
SB-5200DT 型可控温超声波清洗器,宁波新 芝生物科技股份有限公司;
DT5-1 型低速台式离心机,北京医用离心机 厂;
AL204-IC/02 型电子天平(精确至 0.1mg),
METTLER TOLEDO 公司;
UWave-2000微波萃取仪,上海新仪微波化学 科技有限公司
1.2 主要试剂和材料
甲苯(色谱纯),西陇科学股份有限公司;
18 种 PAHs 有证混合标准溶液,100μg/mL;
北京曼哈格生物科技有限公司
八 氘 代 萘 溶 液( 萘 -D8)0.5mg/mL( 美 国 AccuStandard 公司);
十 氘 代 芘 溶 液( 芘 -D10)0.5mg/mL( 美 国 AccuStandard 公司);
十二氘代苝溶液(苝-D12)100μg/mL,北京曼 哈格生物科技有限公司;
高纯氦气(纯度≥99.999%,茂名市民兴气体 有限公司);
PTFE φ13 mm,孔径 0.22 μm 针式过滤器, 市售
炭黑样品,茂名环星新材料股份有限公司
2 检测过程中注意事项及分析结果的影响 因素
2.1 仪器状态
仪器开机后,需要稳定4小时以上,确保仪器 处于正常稳定的状态,仪器运行自动调谐。笔者采 用同一样品,在开机不自动调谐和开机自动调谐两
种状态下进行重复性测试,结果表明,两种状态下 其检测结果相差>10%。因此,建议仪器每周运行 一次自动调谐。调谐实际上就是进行仪器的校准,
通过调节离子源、质量分析器、检测器等各个参 数,使相应的参数达到正常,质谱检测时候得到正 确的质谱图[6]。每次应仔细查看调谐报告中的信
息,若出现异常应根据仪器操作说明进行处理。
其次应每天检查仪器气体流量、钢瓶气压等, 避免仪器系统出现漏气以及中途换气,影响分析 检测的准确度。
2.2 选用合适的进样针
一般来说,同种规格的国产进样针其针尖比 岛津和安捷伦的进样针尖长。进样针的长短对样 品的响应、峰型、重现性都会有一定的影响。针尖
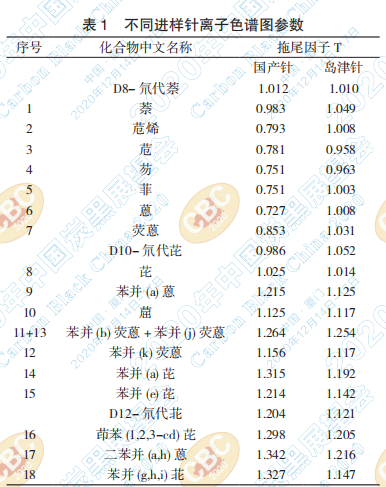
过长,容易造成峰出现拖尾,进而影响峰面积的定 量。一般来说色谱峰的拖尾因子 T 在 0.95~1.05 之 间,则该峰不拖尾。从表1中可以看出,使用国产
的针其色谱峰的拖尾因子误差相对较大。此外, 如果进样针尖过长,穿过了衬管中的石英棉,长期 还可能会会导致堵塞出口、出现鬼峰等现象。若
使用针尖过长的进样针,可以使用隔垫穿过针尖 进行定位,保持进样针的针尖与原装的针尖其长 度一致。
2.3 提取
微波提取法具有操作简便、经济、省时等优 点,同时还可以提高收率[8]。微波提取的操作步骤 如下:
称量炭黑样品→按标准的规定加入溶剂→微 波辐照 20min →过滤→滤液
本文同时在炭黑样品待提取液中添加浓度为 50ng/mL 的 PAHs 标准工作溶液,分别使用微波法 和超声法进行提取炭黑中的 PAHs,比较提取后两
种试液中 18 种 PAHs 的加标回收率,结果见表 2。
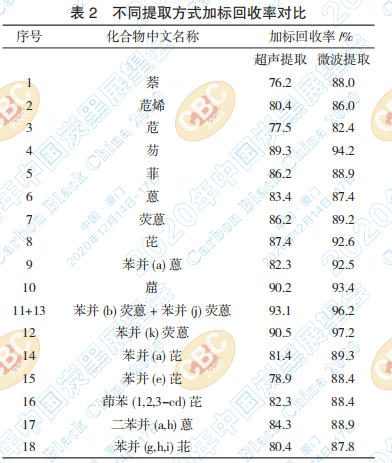
从表2的数据可以看出,超声提取法其加标回 收率为 76~93%,而微波提取法的加标回收率为 82~97%,微波提取法的加标回收率略高,并且在
操作上省时简便。
2.4 净化
标准中指出“使用聚四氟乙烯滤膜过滤后直接 进样”。笔者建议,过滤前应先将提取液经1500r/min 高速离心机离心2 min。该步骤的离心过程相当重
要,滤液的是否净化直接影响到仪器的分析准确 性。纯净度稍低的样液进样后会导致衬管壁受到 污染,当衬管里的污染物积累到一定程度时,会吸
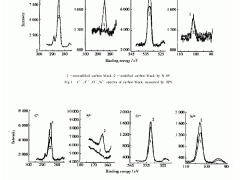
附样品造成峰拖尾/分裂或出现鬼峰,影响分析的 准确度,如图 1 所示的“鬼峰”。

2.5 色谱柱的选用
标准规定的定量方法是内标法。内标法最大 的优点是定量结果与进样量重复性无关,可以有 效避免手动进样体积的误差而引起较大的误差。
使用内标法需要注意的是,内标物需要和待测物 完全分离。一般来说,相邻两个峰其分离度R≥1.5 时,则说明两个峰完全分离;当 R=1.0 时,说明两
个峰刚好分离;当 R < 1.0 时,说明两峰有部分重 叠[7]。若内标物与待测物分离效果不佳,就会对内 标物的峰面积计算产生一定的影响,进而影响了
分析的结果。标准中规定可选用PAHs专用色谱柱 或者固定液化学组成相当的色谱柱。使用两种色 谱柱,PAHs 的分离效果稍微有差别,其分离度见 下表 3。
从表 3 的分离度 R 结果可以看出,选用 PAHs 专用色谱柱各相邻物质的分离度要比普通柱的分 离度稍高,分离效果较佳,因此定量更加准确。图 2
是选用 PAHs 专用色谱柱分离 18 种 PAHs 的离子 色谱图。


3 结 语
本文结合实际,从仪器状态、进样针选用、色 谱柱的选用以及提取分离分析过程等方面分析总 结了炭黑多环芳烃含量检测中可能出现的影响因
素,并针对这些影响因素,总结出行之有效经验对 策,有助于提高分析结果的准确度,确保检测结果 的有效性。
参考文献
[1] 安森萌.超声萃取-高效液相色谱法测定大气PM2.5中 16种多环芳烃[J].环境与发展,2018,30(12).
[2] 蔡素婷,杨三明,郭志顺.气相色谱串联质量法测定水 中的多环芳烃[J]三峡环境与生态,2012,34(3).
[3] 宋世杰,黄韬,周胜,等.博斯腾湖流域沉积物中多环 芳烃的时空分布、来源及生态风险评价[J].环境科学 学报,2019,39(8).
[4] 张朝辉,高帅鹏,王琴琴,等.气相色谱质谱法测定土 壤中16种多环芳烃[J]环境与发展,2019,6.
[5] 谢忠麟.多环芳烃与橡胶制品[J].橡胶工业,2011, 58(6):359-376.
[6] 盛龙生,苏焕华,郭丹滨.色谱质谱联用技术[M].北京: 化学工业出版社,2006
[7] 田丹碧主编.仪器分析[M].北京:化学工业出版社, 2004.
[8] 贾俊强,吴琼英.微波萃取技术的应用[J].食品科技, 2006,(7):5.